- Vận động khó khăn
- Yếu cơ từ gần tới xa
- Giảm trương lực cơ
- Mất hoặc giảm phản xạ
- Rung cơ lưỡi cục bộ
- 50% khả năng sinh con là người lành mang gen bệnh (dị hợp tử)
- 25% khả năng sinh con là người khỏe mạnh không mang gen bệnh
- 25% khả năng sinh con mắc bệnh SMA nặng (đồng hợp tử gen bệnh)
- Triệu chứng và dấu hiệu lâm sàng
- Xét nghiệm men cơ CPK (creatinkinase), sinh thiết cơ, điện tâm đồ tiềm năng EMG
- Xét nghiệm đột biến gen SMN1, xác định đột biến xoá đoạn exon 7 và exon 8
Xét nghiệm gen gây bệnh teo cơ tủy SMA
ThS. Trần Nguyễn An Phú
Khoa Xét nghiệm Di truyền Y học
Bệnh teo cơ tủy (tên tiếng Anh là Spinal Muscular Atrophy - SMA) là bệnh thần kinh – cơ, di truyền lặn trên nhiễm sắc thể số 5, với đặc điểm suy yếu các cơ gốc chi đối xứng do thoái hoá tuần tiến của các tế bào sừng trước tuỷ sống. Hiện tại trên thế giới, tần suất mắc bệnh là 1/10.000 và tần suất người mang gen bệnh là 1/50.
Lâm sàng
Nghĩ đến SMA khi bệnh nhân có các triệu chứng và dấu hiệu sau đây:
SMA được chia thành 5 type dựa vào tuổi khởi phát bệnh và mức độ nặng của bệnh.
|
Thể bệnh |
Khởi phát |
Tuổi thọ |
Vận động |
Triệu chứng |
|
SMA 0 |
Trước sinh |
< 6 tháng |
Không |
Giảm trương lực cơ sơ sinh nặng Suy hô hấp sớm, liệt mặt Trẻ có thể tử vong do suy hô hấp |
|
SMA I (Bệnh Werdnig-Hoffmann) |
< 6 tháng |
Thường ≤ 2 tuổi |
Không thể tự ngồi |
Phản xạ gân xương yếu hoặc mất Giảm trương lực cơ toàn thân nặng Yếu cơ mặt, khó thở, khó nuốt, co rút cơ cục bộ Trẻ có thể tử vong do suy hô hấp và các biến chứng của liệt cơ. |
|
SMA II (Bệnh Dubowitz) |
6-18 tháng |
70% sống đến 25 tuổi |
Có thể tự ngồi được |
Run ngón tay Giảm trương lực cơ Chậm phát triển vận động, không đứng và đi được Biến chứng cong vẹo cột sống và nuốt khó |
|
SMA III (Bệnh Kugelberg-Welander) |
> 18 tháng |
Bình thường |
Có thể tự đi được |
Chậm phát triển vận động Yếu cơ gốc chi, co rút cơ, cong vẹo cột sống |
|
SMA IV |
Trưởng thành |
Bình thường |
Bình thường |
|
Nguyên nhân di truyền
Nguyên nhân gây bệnh SMA là do đột biến gen SMN (survival monitor neuron) trên nhiễm sắc thể số 5 (5q13). Gen SMN quy định tổng hợp protein SMN dài 294 axít amin hiện diện chủ yếu ở các tế bào thần kinh vận động tủy sống. Đột biến ở gen SMN khiến cho các tế bào thần kinh vận động trong tủy sống và não bộ không hoạt động.
Gen SMN có 9 exon (exon 1, 2a và 2b, 3 - 8) với 2 phiên bản rất giống nhau là SMN1 (SMNt) và SMN2 (SMNc), chỉ khác nhau ở một vài nucleotide (hình 1). Phiên bản SMN1 tổng hợp được protein có chức năng chủ yếu, trong khi protein SMN2 có rất ít chức năng. Đồng hợp tử đột biến xoá mất exon 7 và 8 gen SMN1 gây ra bệnh ở 95% các trường hợp hợp SMA nặng. Chỉ khoảng 5% bệnh nhân SMA là do đột biến điểm trên gen SMN1.
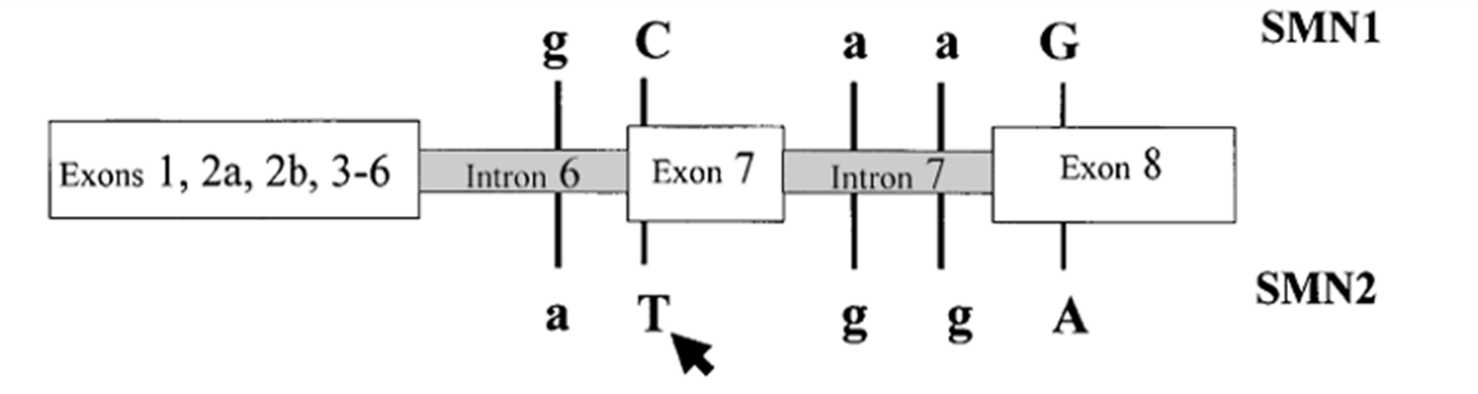
Hình 1. Sơ đồ gen SMN và 5 vị trí nucleotide khác nhau giữa gen SMN1 và SMN2
Cơ chế di truyền
Gen gây bệnh di truyền lặn trên nhiễm sắc thể thường số 5 nên tỉ lệ mắc bệnh là như nhau ở cả hai giới nam và nữ. Người mang 1 gen bệnh (dị hợp tử) sẽ không biểu hiện bệnh nhưng có thể có con bị bệnh nặng nếu lấy người có cùng loại đột biến gen.
Trường hợp bố và mẹ đều là người mang gen bệnh, nguy cơ sinh con bị bệnh mỗi lần mang thai như sau (hình 2):
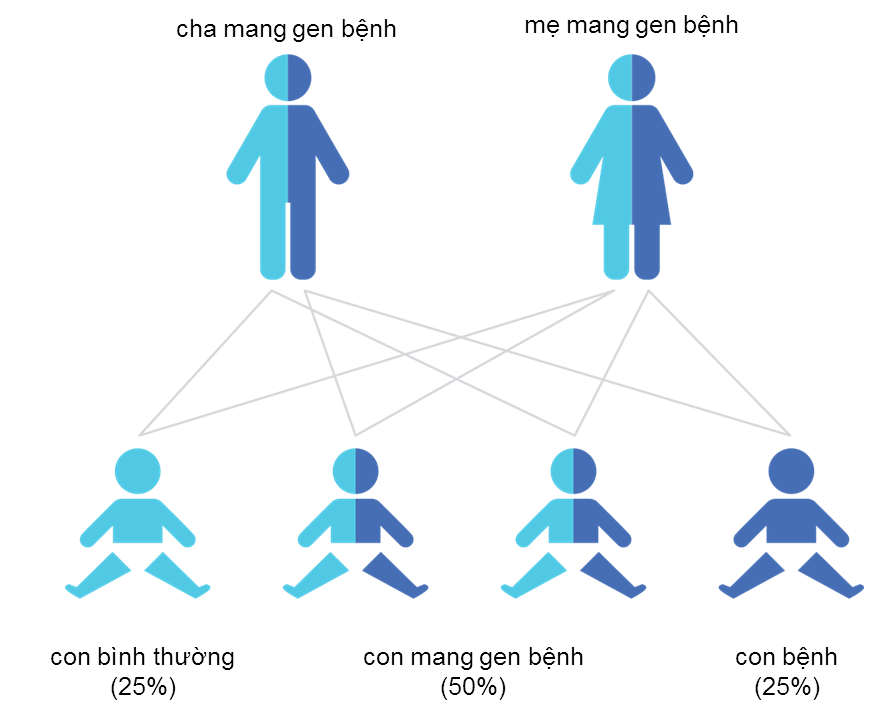
Hình 2. Sơ đồ di truyền gen gây bệnh SMA
Chẩn đoán
Chẩn đoán bệnh dựa trên:
Điều trị
Hiện tại phương pháp chủ yếu là điều trị triệu chứng, phòng ngừa các biến chứng và hỗ trợ chức năng sống cho người bệnh.
Các nhà khoa học trên thế giới vẫn đang nghiên cứu thử nghiệm các phương pháp điều trị đặc hiệu, trong đó có phương pháp điều trị gen.
Phòng ngừa
Phương pháp phòng ngừa bệnh hiệu quả nhất là tư vấn di truyền, xét nghiệm tầm soát đột biến gen SMN cho các cặp đôi hoặc xét nghiệm chẩn đoán trước sinh đột biến gen SMN cho thai của các cặp vợ chồng đã có tiền sử sinh con bị bệnh SMA.
Liên hệ xét nghiệm
Khoa Xét nghiệm Di truyền Y học – Bệnh viện Từ Dũ
191 Cống Quỳnh, Quận 1, TPHCM; Khu N – Lầu 3
ĐT: 028 5404 2829 – 239 hoặc số trực tiếp: 028 5404 2812
Tài liệu tham khảo
Clermont O, Burlet P, Benit P, Chanterau D, Saugier‐Veber P, Munnich A, et al (2004). Molecular analysis of SMA patients without homozygous SMN1 deletions using a new strategy for identification of SMN1 subtle mutations. Hum Mutat, 24: 417‐427.
Cusin V, Clermont O, Chantereau D, Elion J (2003). Prevalence of SMN1 deletion and duplication in carrier and normal populations: implication for genetic counselling. JMed Genet, 40, e39.
Kết nối với Bệnh viện Từ Dũ


